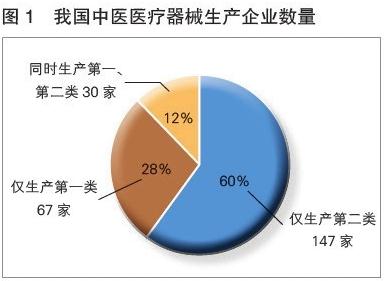
醫療器械是守護人類健康的重要工具,其安全性與有效性直接關系到使用者的生命健康。為確保產品在上市前達到規定的質量標準,各國監管機構都設立了嚴格的審批與監管流程。在我國,醫療器械根據風險程度實行分類管理,其中第一類醫療器械風險程度最低,實行備案管理。即便如此,其上市前仍需跨越一系列關鍵環節,確保“測探高質量,試出新未來”。
一、 產品分類與路徑確認
首要且關鍵的一步是準確界定產品屬于第一類醫療器械。企業需根據國家藥品監督管理局發布的《醫療器械分類目錄》,結合產品的預期用途、結構特征、使用形式等,進行專業判定。確認屬于第一類后,便明確了上市前的管理方式為“備案”,而非風險更高的第二、三類產品的“注冊”審批。這一步是后續所有工作的基石,分類錯誤將導致整個合規路徑的偏差。
二、 產品研制與性能驗證
在正式備案前,企業必須完成產品的研制工作,并確保其滿足基本的安全有效性要求。這包括:
- 設計開發:依據醫療器械生產質量管理規范(GMP)的要求,建立并實施設計開發控制程序,保留完整的研發記錄。
- 產品檢驗:產品必須符合強制性國家標準(GB)或行業標準(YY)。若無國行標,企業需自行制定產品技術要求。企業需根據技術要求,對產品進行全性能自檢,或委托有資質的醫療器械檢驗機構出具產品檢驗報告。檢驗項目通常包括物理性能、化學性能、生物安全性(若適用)等。
- 臨床評價(如適用):對于第一類醫療器械,通常免于進行臨床試驗。但企業仍需通過文獻資料、同類產品對比等方式進行臨床評價,證明產品滿足基本的安全有效性原則。
三、 質量管理體系建立
生產企業必須建立符合《醫療器械生產質量管理規范》及其附錄要求的質量管理體系,這是保障產品持續穩定上市的基礎。體系需覆蓋設計開發、采購、生產、質量控制、銷售及售后服務等全過程。對于第一類醫療器械,監管方會通過事后監管、現場檢查等方式對企業體系運行情況進行核查。
四、 準備并提交備案資料
這是正式向監管部門申報的核心步驟。企業需向所在地的市級藥品監督管理部門(具體層級依地方規定)提交完整的備案資料,主要包括:
- 第一類醫療器械備案表。
- 安全風險分析報告:概述產品主要風險及控制措施。
- 產品技術要求:詳細規定產品的性能指標和檢驗方法。
- 產品檢驗報告:證明產品符合技術要求的證據。
- 臨床評價資料(如適用)。
- 產品說明書及最小銷售單元標簽設計樣稿。
- 生產制造信息:包括生產工藝簡述、生產場地信息等。
- 證明性文件:如營業執照副本、企業符合生產質量管理規范的自查報告等。
- 符合性聲明:聲明所提交資料真實合規,產品屬于第一類醫療器械。
五、 監管部門備案與公示
藥品監督管理部門在收到備案資料后,對資料的完整性、規范性進行審核。對于備案資料符合要求的,監管部門將予以備案,并在其官方網站上公示備案信息(包括產品名稱、備案人名稱、備案號等),備案號格式通常為“×械備××××××××”。此時,產品即獲得上市許可。
六、 后續生產與上市后監管
獲得備案憑證并不意味著監管的結束,而是新階段的開始。企業必須:
- 嚴格按備案技術要求組織生產,任何影響安全有效的變更都需進行變更備案或報告。
- 履行上市后監測義務,收集不良事件信息并及時報告。
- 接受監管部門的各類監督檢查,確保質量管理體系持續有效運行。
****
對于第一類醫療器械,“備案制”簡化了上市前的行政審批流程,但絕不意味著質量要求的降低。“測探高質量”貫穿于從研發設計到生產放行的每一個環節;“試出新未來”則體現在通過嚴謹的合規流程,將安全可靠的產品推向市場,最終贏得用戶信任并實現商業價值。企業唯有深刻理解并嚴格落實每一步要求,方能行穩致遠,在保障公眾用械安全的擁抱產業發展的新未來。